
Lab work is divided into three modules. This page presents details only for the first two modules. Details of the third module are not available in OpenCourseWare, because the module is based on recent research that is being prepared for formal publication.
For modules 1 and 2, each lab session is presented on a linked web page with an introduction, protocol, and reagents list, followed by a “for next time” homework assignment.
Lab Basics
- General dos and don’ts of working in the lab.
Guidelines for Maintaining Your Lab Notebook
- How to maintain a good lab notebook.
Guidelines for Working in the Tissue Culture Facility
- Procedures for doing tissue culture work.
Orientation Session (First Class)
Lecturer: Dr. Natalie Kuldell
There are six stations for you and your lab partner to visit on this lab tour. Some will be guided tours with a TA or faculty there to help you and others are self-guided, leaving you and your partner to try things on your own. Your visit to each station will last 10-15 minutes. It doesn’t matter which station you visit first but you must visit them all before you leave today. Your lab practical next time will assess your mastery of each station.
Day 1: Lab Tour
Module 1: Genome Engineering
Instructors: Prof. Drew Endy, Dr. Natalie Kuldell, and Dr. Agi Stachowiak
Lecturer: Prof. Drew Endy
TA: Laure-Anne Ventouras
In this experiment, we will consider the genome of a virus, namely the bacteriophage M13. M13 is a self-assembling nano-machine with a compact genome that has been optimized by evolution to commandeer its bacterial host. Approximately 1000 new viruses are generated from a single infection event. Imagine harnessing this production. What could we build and what natural processes could we better understand? One approach we’ll take is to modify the existing genome in a subtle but useful way, namely by adding a useful sequence-tag that modifies the bacteriophage coat. We’ll examine how this modification affects the coat protein’s expression and overall phage production. Another approach we’ll take is to start from scratch, undertaking a full throttle redesign of the bacteriophage genome. We’ll employ a commercial DNA synthesis company to compile the redesigned genomic program and then we’ll see if it encoded infective M13 and if the genome of the bacterial host affects bacteriophage production. Through these investigations we’ll ask: is nature’s M13 genome “perfect” or can we do better?
Photo of M13-coated coli removed due to copyright restrictions.
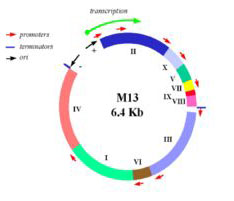
Map of M13 genome from M. Blaber. (Images courtesy of Dr. Michael Blaber. Used with permission.)
| MODULES DAY | LAB TOPICS |
|---|---|
| 1 | Start-up genome engineering |
| 2 | Agarose gel electrophoresis |
| 3 | DNA ligation and bacterial transformation |
| 4 | Examine candidate clones |
| 5 | M13.1 |
| 6 | Western analysis |
| 7 |
Lecture on environmental health and safety (no materials) |
| 8 | Oral presentations |
Working Page for M13 Refactoring
Pedagogy
Module 2: Expression Engineering
Instructors: Dr. Natalie Kuldell and Dr. Agi Stachowiak
Lecturer: Dr. Natalie Kuldell
TA: Alice Lo
In this experiment, we will consider unintended and unpredicted effects of an experimental perturbation. Our goal is a precise one, namely to silence gene expression of a measurable gene, luciferase, using RNA interference (RNAi). Each group will begin by designing a short interfering RNA (siRNA) against luciferase, but as we’ll see, siRNAs can vary in efficacy and specificity. After transfecting a mammalian cell line with the siRNA you’ve designed and a reporter plasmid, we will evaluate the silencing using a luciferase assay and microarray technology. The first assay evaluates the efficacy of the siRNA in silencing. The second assay gives genome-wide expression data to reveal the specificity of your siRNA for the gene you’ve targeted. Through this combined approach, we’ll assess the balance of targeted and off-target effects.

Child jumping through sprinkler, with sunlight. Photo by Natalie Kuldell.
| MODULES DAY | LAB TOPICS |
|---|---|
| 1 | siRNA design and introduction to cell culture |
| 2 | Transfection |
| 3 | Luciferase assays and RNA prep |
| 4 | Journal article discussion |
| 5 | cDNA synthesis and microarray |
| 6 | Microarray data analysis |
| 7 | High throughput technologies (no materials) |
| 8 | Oral presentations |
Pedagogy
Module 3: Biomaterials Engineering
“Invention” is a wonderful word, derived from words meaning “scheme” and “a finding out.” Inventors draw on materials provided by the natural world, refining and combining them in insightful ways, to make something useful. In this experimental module we will invent materials by manipulating biological systems, namely the bacteriophage M13. We will use a very slightly modified phage to build Iridium nanowires that we’ll visualize on the transmission electron microscope. Then we’ll let the phage themselves do the building, making an electrochromic device that’s both fun and potentially useful. Drawing on the rich stockroom of biological elements and a good but incomplete understanding of their behavior, we’ll hope to invent some novel materials with real-world applications.

TEM of M13E4 after CoCl2/NaBH4 treatment, image courtesy of Natalie Kuldell, Anthony Garratt-Reed and KiTae Nam.
Details of this module are not available in OCW, because the module is based on recent research that is on the way to formal publication.







